Introduction to ML-UMR: Multilevel Unanchored Meta-Regression
Ahmad Sofi-Mahmudi
2026-02-06
Source:vignettes/mlumr_tutorial.Rmd
mlumr_tutorial.RmdIntroduction
The mlumr package implements Multilevel
Unanchored Meta-Regression (ML-UMR) for population-adjusted
indirect comparisons in disconnected networks. This tutorial will guide
you through the complete workflow from data preparation to model fitting
and interpretation.
When to Use ML-UMR
ML-UMR is appropriate when:
- You have individual patient data (IPD) for one treatment (the “index” treatment)
- You have only aggregate data (AgD) for another treatment (the “comparator” treatment)
- There is no common comparator linking the two studies (unanchored comparison)
- There are population imbalances in prognostic factors between studies
- You want to perform population-adjusted indirect comparisons
Installation
# Install dependencies
install.packages(c("cmdstanr", "dplyr", "tidyr", "ggplot2", "posterior",
"randtoolbox", "copula", "tibble", "rlang", "purrr", "glue"))
# Install CmdStan
cmdstanr::install_cmdstan()
# Install mlumr
devtools::install_local("path/to/mlumr")Example: Simulated Data
For this tutorial, we’ll use simulated data to demonstrate the ML-UMR workflow. In practice, you would use your own IPD and AgD.
Generate Simulation Data
Let’s generate data for an unanchored comparison with two prognostic factors:
set.seed(123)
# Scenario where SPFA holds (no effect modification)
data <- generate_simulation_data(
n_index = 200, # Sample size for index treatment (IPD)
n_comparator = 150, # Sample size for comparator treatment (AgD)
n_covariates = 2, # Number of prognostic factors
effect_modification = "none", # SPFA holds
population_shift = 0.5, # Imbalance between populations
correlation = 0.3, # Correlation between covariates
seed = 123
)
# View the data structure
print(data)
#> ML-UMR Simulation Data
#> ======================
#>
#> Design:
#> Likelihood: binomial
#> Index treatment (IPD): n = 200
#> Events: 82
#> Comparator treatment (AgD): 1 subgroups/studies
#> Total n = 150 , Total events = 104
#> Number of covariates: 2
#> Effect modification: none
#>
#> True Parameters:
#> mu_index = -0.5
#> mu_comparator = -0.3
#> beta_index: 0.5, 1
#> beta_comparator: 0.5, 1
#>
#> True Marginal Effects:
#> LOR (index pop): -0.157
#> LOR (comparator pop): -0.153
#> RD (index pop): -0.038
#> RD (comparator pop): -0.037The simulated data includes:
- IPD: Individual-level data with binary outcome and covariates
- AgD: Aggregate summary statistics (sample size, events, covariate means/SDs)
- Truth: True parameter values (for validation)
Step 1: Set Up IPD
First, we prepare the individual patient data from the index treatment:
ipd <- set_ipd_unanchored(
data = data$ipd,
treatment = "treatment",
outcome = "outcome",
covariates = c("x1", "x2")
)
print(ipd)
#> $data
#> # A tibble: 200 × 5
#> .study .trt .outcome x1 x2
#> <chr> <chr> <int> <dbl> <dbl>
#> 1 IPD_Study Treatment_A 1 -0.560 1.93
#> 2 IPD_Study Treatment_A 1 -0.230 1.18
#> 3 IPD_Study Treatment_A 1 1.56 0.215
#> 4 IPD_Study Treatment_A 1 0.0705 0.539
#> 5 IPD_Study Treatment_A 0 0.129 -0.356
#> 6 IPD_Study Treatment_A 0 1.72 0.0602
#> 7 IPD_Study Treatment_A 0 0.461 -0.614
#> 8 IPD_Study Treatment_A 0 -1.27 -0.947
#> 9 IPD_Study Treatment_A 0 -0.687 1.37
#> 10 IPD_Study Treatment_A 0 -0.446 -0.185
#> # ℹ 190 more rows
#>
#> $n
#> [1] 200
#>
#> $n_events
#> [1] 82
#>
#> $treatment
#> [1] "Treatment_A"
#>
#> $covariates
#> [1] "x1" "x2"
#>
#> $likelihood
#> [1] "binomial"
#>
#> $type
#> [1] "ipd"
#>
#> attr(,"class")
#> [1] "ipd_unanchored" "list"The IPD object contains:
- Standardized data structure
- Sample size and event counts
- Covariate names
Step 2: Set Up AgD
Next, we prepare the aggregate data from the comparator treatment:
agd <- set_agd_unanchored(
data = data$agd,
treatment = "treatment",
outcome_n = "n_total",
outcome_r = "n_events",
cov_means = c("x1_mean", "x2_mean"),
cov_sds = c("x1_sd", "x2_sd"),
cov_types = c("continuous", "continuous")
)
print(agd)
#> $data
#> # A tibble: 1 × 8
#> .study .trt .n .r x1_mean x1_sd x2_mean x2_sd
#> <chr> <chr> <dbl> <int> <dbl> <dbl> <dbl> <dbl>
#> 1 AgD_Study Treatment_B 150 104 0.418 0.966 0.626 1.04
#>
#> $n
#> [1] 150
#>
#> $n_events
#> [1] 104
#>
#> $treatment
#> [1] "Treatment_B"
#>
#> $covariates
#> [1] "x1" "x2"
#>
#> $cov_info
#> $cov_info$x1
#> $cov_info$x1$mean_col
#> [1] "x1_mean"
#>
#> $cov_info$x1$sd_col
#> [1] "x1_sd"
#>
#> $cov_info$x1$type
#> [1] "continuous"
#>
#>
#> $cov_info$x2
#> $cov_info$x2$mean_col
#> [1] "x2_mean"
#>
#> $cov_info$x2$sd_col
#> [1] "x2_sd"
#>
#> $cov_info$x2$type
#> [1] "continuous"
#>
#>
#>
#> $likelihood
#> [1] "binomial"
#>
#> $type
#> [1] "agd"
#>
#> attr(,"class")
#> [1] "agd_unanchored" "list"For binary covariates, set cov_sds = NA and
cov_types = "binary".
Step 3: Combine IPD and AgD
Combine the two data sources into a single network object:
network <- combine_unanchored(ipd, agd)
print(network)
#> Unanchored Comparison Data
#> ===========================
#>
#> Likelihood: binomial
#>
#> Index treatment (IPD): Treatment_A
#> - N = 200
#> - Events = 82 (41.0%)
#>
#> Comparator treatment (AgD): Treatment_B
#> - N = 150
#> - Events = 104 (69.3%)
#>
#> Covariates ( 2 ): x1, x2
#>
#> Integration points: Not yet added (use add_integration_unanchored())The package checks that:
- Covariate names match between IPD and AgD
- Both datasets are properly formatted
Step 4: Add Integration Points
ML-UMR uses quasi-Monte Carlo integration to handle aggregate data. We generate integration points using Sobol sequences and a Gaussian copula to preserve correlations:
network <- add_integration_unanchored(
network,
n_int = 64, # Number of integration points (powers of 2 recommended)
x1 = distr(qnorm, mean = x1_mean, sd = x1_sd),
x2 = distr(qnorm, mean = x2_mean, sd = x2_sd)
)
#> Computing correlation matrix from IPD...
#> Added 64 integration points for AgD
print(network)
#> Unanchored Comparison Data
#> ===========================
#>
#> Likelihood: binomial
#>
#> Index treatment (IPD): Treatment_A
#> - N = 200
#> - Events = 82 (41.0%)
#>
#> Comparator treatment (AgD): Treatment_B
#> - N = 150
#> - Events = 104 (69.3%)
#>
#> Covariates ( 2 ): x1, x2
#>
#> Integration points: 64 (QMC with Gaussian copula)The distr() function specifies the marginal distribution
for each covariate. Common distributions:
-
Continuous:
distr(qnorm, mean = ..., sd = ...) -
Binary:
distr(qbern, prob = ...) -
Other: Any quantile function (e.g.,
qgamma,qbeta)
Visualize Covariate Distributions
Compare the covariate distributions between populations:
p1 <- plot_covariate_distribution(network, "x1")
p2 <- plot_covariate_distribution(network, "x2")
print(p1)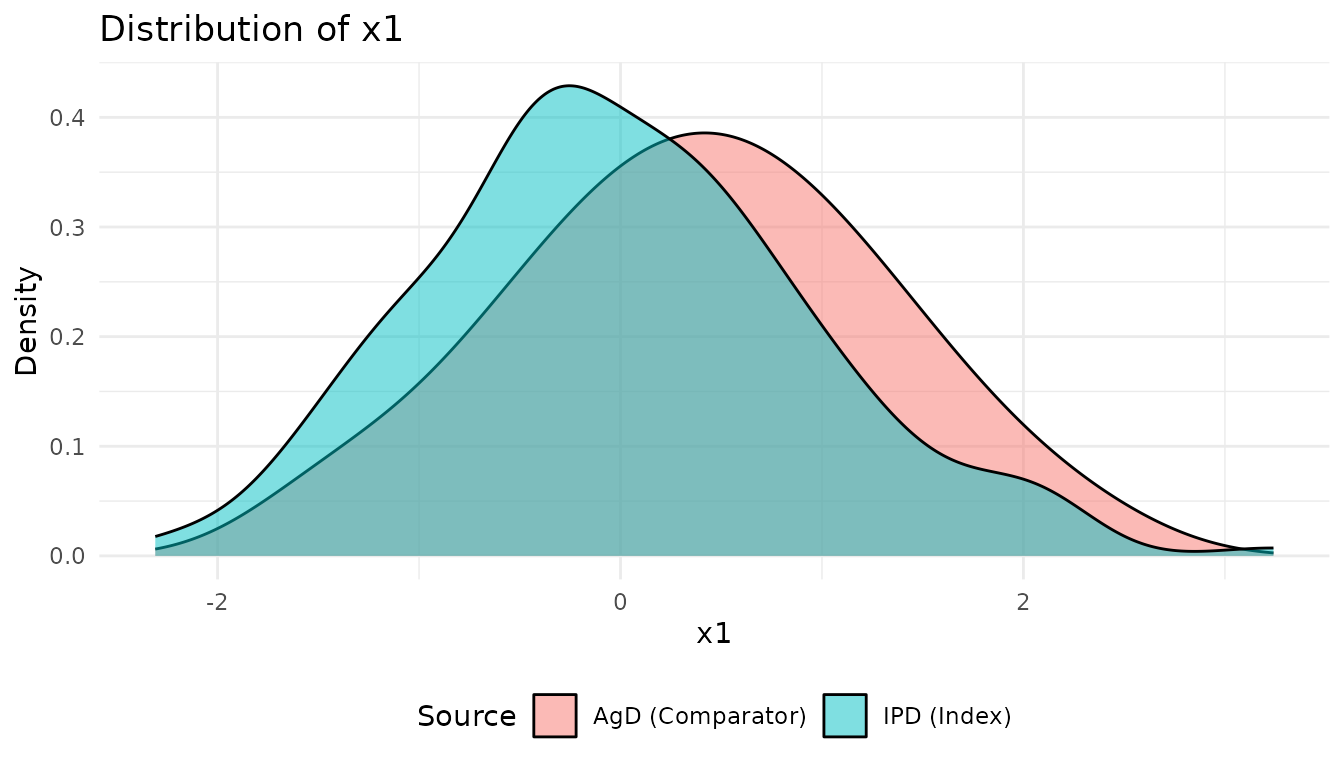
print(p2)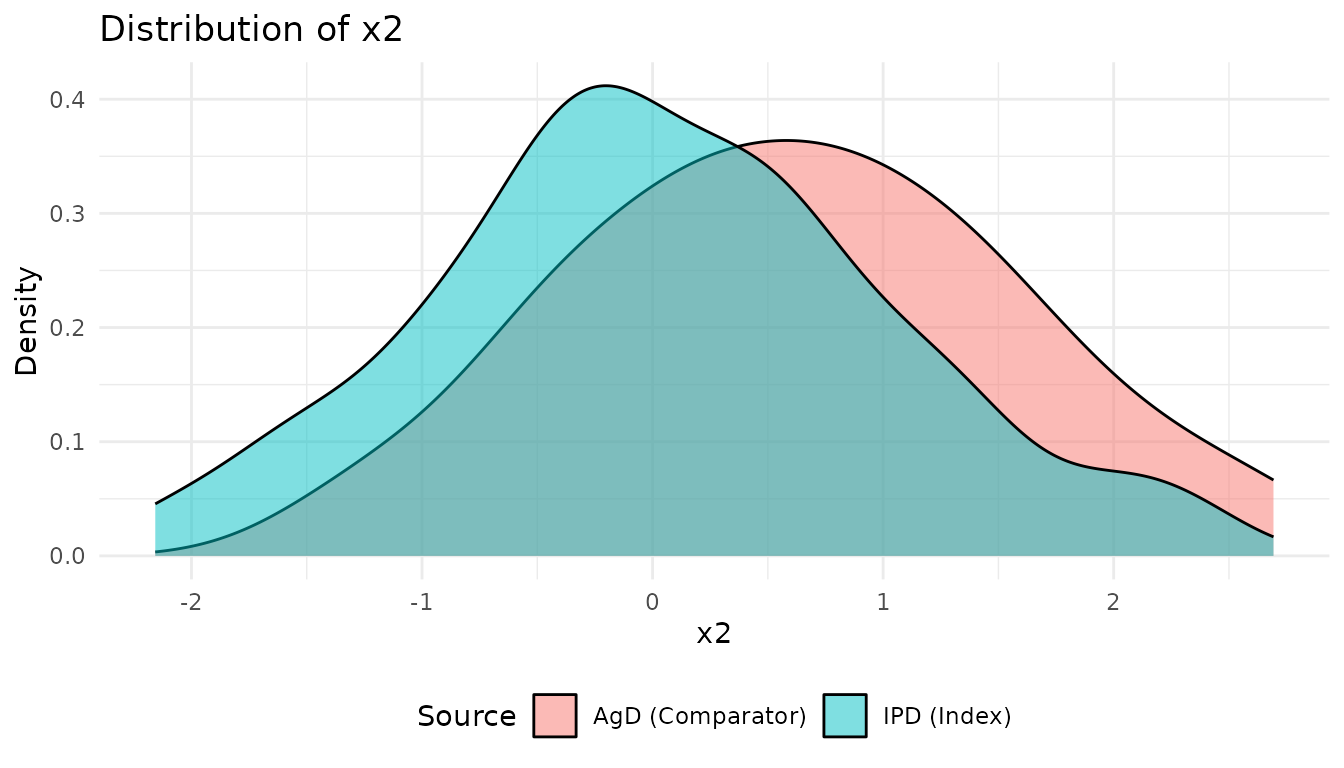
These plots show the population imbalance that ML-UMR will adjust for.
Step 5: Fit ML-UMR Model
Model with SPFA
First, fit a model that assumes the Shared Prognostic Factor Assumption (SPFA), meaning prognostic factors have the same effect on outcomes for both treatments:
fit_spfa <- mlumr(
data = network,
spfa = TRUE, # Invoke SPFA
prior_intercept = prior_normal(0, 10),
prior_beta = prior_normal(0, 10),
iter_warmup = 1000,
iter_sampling = 2000,
chains = 4,
seed = 123
)
print(fit_spfa)Step 6: Model Comparison
Compare the models using the Deviance Information Criterion (DIC):
# Calculate DIC for both models
dic_spfa <- calculate_dic(fit_spfa)
dic_relaxed <- calculate_dic(fit_relaxed)
# Compare
comparison <- compare_models(fit_spfa, fit_relaxed)Interpretation:
- Lower DIC indicates better fit
- ΔDIC > 5 suggests meaningful difference
- If SPFA model has lower DIC, assume shared prognostic factors
- If Relaxed model has lower DIC, there is evidence of effect modification
Step 7: Extract Results
Model Summary
summary(fit_spfa)This provides:
- MCMC diagnostics (Rhat, ESS)
- Intercept estimates
- Regression coefficients
- Marginal treatment effects
Marginal Treatment Effects
Extract marginal effects in both populations:
effects <- marginal_effects(fit_spfa, population = "both")
print(effects)The output includes:
- LOR: Log odds ratio (index vs comparator)
- RD: Risk difference (index - comparator)
- RR: Risk ratio (index / comparator)
In both:
- Index population: Effect if comparator were given to IPD population
- Comparator population: Effect if index were given to AgD population
Step 8: Visualizations
Marginal Effects Plot
plot(fit_spfa, type = "marginal")Forest Plots
# Log odds ratio
plot_marginal_effects_forest(fit_spfa, effect_type = "lor")
# Risk difference
plot_marginal_effects_forest(fit_spfa, effect_type = "rd")
# Risk ratio
plot_marginal_effects_forest(fit_spfa, effect_type = "rr")Validation with Simulated Data
Since we used simulated data, we can compare estimates to true values:
Effect Modification Example
Let’s demonstrate what happens when there is strong effect modification:
# Generate data with strong effect modification
data_em <- generate_simulation_data(
n_index = 200,
n_comparator = 150,
n_covariates = 2,
effect_modification = "strong",
seed = 456
)
# Set up and fit both models
# [Code to set up IPD, AgD, network, and integration points]
# Fit SPFA model
fit_em_spfa <- mlumr(data = network_em, spfa = TRUE, seed = 789)
# Fit Relaxed model
fit_em_relaxed <- mlumr(data = network_em, spfa = FALSE, seed = 789)
# Compare models
compare_models(fit_em_spfa, fit_em_relaxed)
# Extract effect modification parameters
delta_beta <- fit_em_relaxed$summary[grep("^delta_beta",
fit_em_relaxed$summary$variable), ]
print(delta_beta)When effect modification is present:
- Relaxed model will have lower DIC
-
delta_betaparameters show the difference in covariate effects between treatments - Credible intervals for
delta_betaexcluding zero indicate effect modification
Practical Recommendations
Sample Size
- IPD: At least 100-200 patients recommended
- AgD: At least 50-100 patients recommended
- Larger samples provide more precise estimates
Integration Points
- Use powers of 2: 32, 64, 128, 256
- More points = better accuracy but slower computation
- 64 is usually sufficient
- Test with different values to check sensitivity
Priors
- Default:
prior_normal(0, 10)(weakly informative) - Adjust based on prior knowledge:
- Tighter priors:
prior_normal(0, 2.5) - More vague:
prior_normal(0, 20)
- Tighter priors:
Key Assumptions
ML-UMR relies on several assumptions:
No unmeasured confounding: All prognostic factors that differ between populations and modify treatment effects are measured and included
Correct functional form: Linear relationships on logit scale
Correct covariate distributions: Integration points accurately represent AgD population
SPFA (if invoked): Prognostic factors have same effect on outcomes for both treatments
SPFA is testable via model comparison, but unmeasured confounding is not. Careful consideration of what covariates to include is crucial.
Comparison with Other Methods
| Method | Pros | Cons |
|---|---|---|
| MAIC | Simple, widely used | Strong assumptions, efficiency loss, limited to comparator population |
| STC | Flexible regression | Aggregation bias, limited to comparator population |
| ML-UMR | No aggregation bias, multiple populations, tests SPFA | Requires good knowledge of AgD distributions |
Further Resources
- See
?mlumrfor detailed function documentation - See
inst/examples/standalone_example.Rfor complete working example - See package vignettes for advanced topics
Session Information
sessionInfo()
#> R version 4.5.2 (2025-10-31)
#> Platform: x86_64-pc-linux-gnu
#> Running under: Ubuntu 24.04.3 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
#>
#> locale:
#> [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
#> [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
#> [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
#> [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: UTC
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] ggplot2_4.0.2 dplyr_1.2.0 mlumr_0.1.0
#>
#> loaded via a namespace (and not attached):
#> [1] sass_0.4.10 utf8_1.2.6 generics_0.1.4
#> [4] lattice_0.22-7 digest_0.6.39 magrittr_2.0.4
#> [7] evaluate_1.0.5 grid_4.5.2 RColorBrewer_1.1-3
#> [10] mvtnorm_1.3-3 fastmap_1.2.0 jsonlite_2.0.0
#> [13] Matrix_1.7-4 rngWELL_0.10-10 purrr_1.2.1
#> [16] scales_1.4.0 stabledist_0.7-2 randtoolbox_2.0.5
#> [19] numDeriv_2016.8-1.1 textshaping_1.0.4 jquerylib_0.1.4
#> [22] cli_3.6.5 rlang_1.1.7 pspline_1.0-21
#> [25] gsl_2.1-9 withr_3.0.2 cachem_1.1.0
#> [28] yaml_2.3.12 tools_4.5.2 copula_1.1-6
#> [31] vctrs_0.7.1 R6_2.6.1 stats4_4.5.2
#> [34] lifecycle_1.0.5 ADGofTest_0.3 fs_1.6.6
#> [37] ragg_1.5.0 pcaPP_2.0-5 pkgconfig_2.0.3
#> [40] desc_1.4.3 pkgdown_2.2.0 pillar_1.11.1
#> [43] bslib_0.10.0 gtable_0.3.6 glue_1.8.0
#> [46] systemfonts_1.3.1 xfun_0.56 tibble_3.3.1
#> [49] tidyselect_1.2.1 knitr_1.51 farver_2.1.2
#> [52] htmltools_0.5.9 labeling_0.4.3 rmarkdown_2.30
#> [55] compiler_4.5.2 S7_0.2.1References
Chandler, C. & Ishak, J. (2025). “Anchors Away: Navigating Unanchored Indirect Comparisons With Multilevel Unanchored Meta-Regression (ML-UMR).” ISPOR Europe 2025.
Phillippo, D.M., et al. (2020). “Multilevel Network Meta-Regression for population-adjusted treatment comparisons.” Journal of the Royal Statistical Society: Series A, 183(3):1189-1210.